This abstract was presented today, May 1st at the 2018 Association for Research in Vision and Opthalmology (ARVO) meetings in Honolulu, Hawaii by Siamak Yousefi, Hao Chen, Jesse Ingels, Sumana R. Chintalapudi, Megan Mulligan, Bryan W. Jones, Vanessa Marie Morales-Tirado, Pete Williams, Simon W. John, Felix Struebing, Eldon E. Geisert, Monica Jablonski, Lu Lu, Robert Williams
Purpose
We are developing methods to define molecular signatures of cellular stress during early stages of glaucoma for major subtypes of retinal ganglion cells (RGCs). Our first aim is to develop reliable mRNA biomarkers for RGC subtypes in the DBA/2J (D2) mouse model prior to disease onset. Our second objective is to quantify cellular stress in RGC subtypes at early stages of disease using known sets of stress-responsive transcripts (e.g. Struebing et al, 2016 PMID:27733864; Williams et al. 2017, PMID:28209901; Lu et al, ARVO 2018).
Methods
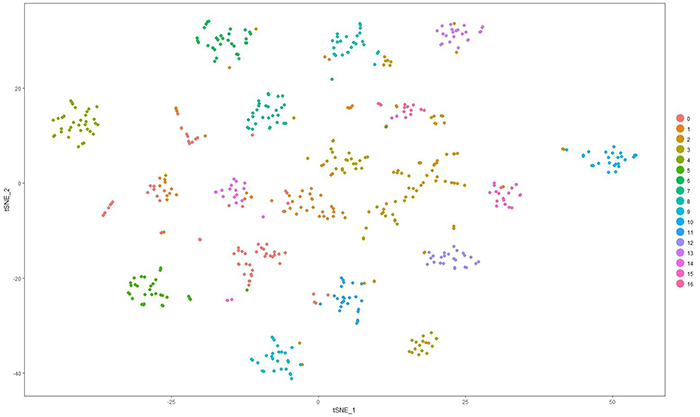
Whole retinas from D2 or D2.Cg-Tg(Thy1-CFP)23Jrs/SjJ at 130 to 150 days-of-age were dissociated gently and size selected (>10 µm). RGCs were enriched using THY1 antibody-coated beads. Fluidigm HT microfluidics plates were used to isolate and generate scRNA-seq libraries of full length polyA-positive mRNAs using SMART-Seq v4. Libraries were sequenced using HiSeq3000, PE151. Following alignment using STAR, expression was normalized to log2(FPKM+1) across ~25,000 unique transcript models. Cells with fewer than 1000 detected genes and genes expressed in fewer than 1% of RGCs were excluded. Sets of genes with high variance and/or high expression were used for principal component analysis (PCA). Twenty PCs were used for graph-based unsupervised clustering and visualized using t-distributed stochastic neighbor embedding (tSNE). Gene specificity was computed for all transcripts across all clusters. The top transcripts per cluster with expression >1 in 1% or more of cells, were used to diagnose cellular identify of clusters. The top 30 genes per cluster were searched in PubMed against a panel of cell and tissue specific terms using Chilibot.
Results
The scRNA-seq protocol generates 150,000 – 200,000 uniquely mapped mRNA reads/cell and ~5000 genes/cells. We currently have 1600 cells, of which over half are RGCs. Around 75% of cells are positive for two or more of the following RGC markers: Thy1, Rbpms, Rbpms2, Jam2, G3bp1, and Ywhaz. This set of cells and different subsets of genes are now being used for RGC clustering. We have identified at least 17 clusters in initial datasets using these protocols and are now linking clusters to major classes of RGCs.
Conclusions
Molecular signatures of cellular stress and RGC subtypes in early stage of glaucoma should now be identifiable using unsupervised learning techniques.